
Bioinformatics
For a thorough study of geobiology (or environmental microbiology) I believe it is vital to combine observations from the field with experiments in the lab. More recently, a third component can be added to the list: sequence analysis, and bioinformatics more broadly. On the “in Situ”, “in Vitro”, and “in Silico” pages of this site, I highlight some of my research and teaching efforts in these three components of geobiology.
This page is showcases some approaches in bioinformatics.
Teaching
BVCN
The Bioinformatics Virtual Coordination Network (BVCN) is a community driven effort
started by Dr. Ben Tully during the shutdown of many research institutes in the United Stated during the COVID-19 pandemic of 2020.
The BVCN provides a slack channel where learners (and instructors) can ask questions of, and discuss approaches/tools with, more than 600 members.
In addition, the BVCN instructors release video tutorials on the BVCN Youtube channel.
Thus far, I have contributed videos on metagenomic binning
and bin evaluation.
These lessons were accompanied by a DAS Tool demonstration
and a checkm tutorial.
Brief History of Metagenomics
I have co-taught a seminar course with Prof. Victoria Orphan,
loosely based on the introduction chapter 1.1 of my PhD thesis.
The course covered conceptual and technological advances leading up to the metagenomics boom of today.
Outlining the course content here would take up a bit too much space, but will be the subject of a separate post, which I’ll link here.
Research
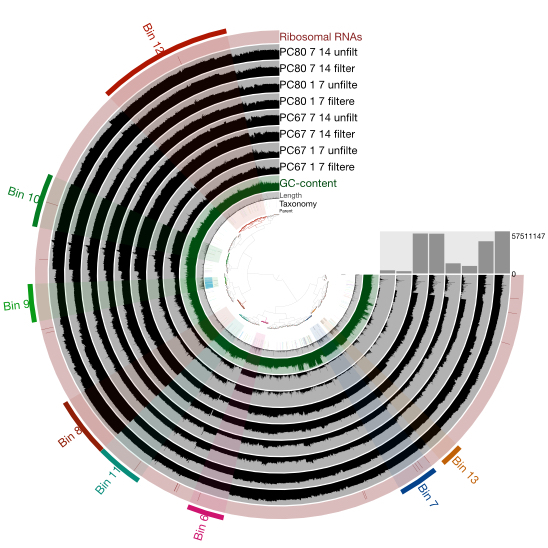
Genomes from Metagenomes
Our ability to recover genomes from environmental samples, through metagenomics and single cell based approaches,
has tranformed our view of microbial diversity.In addition, it has shed new light on the phylogenetic distribution and evolution of microbial metabolisms,
such as methanogenesis. As a graduate student, I was an early adopter of genome resolved metagenomics,
resulting in more genomic information on organisms capable of anaerobic ammonium oxidation (anammox bacteria).
In addition, a community-wide analysis of an anammox reactor in the main line of a Dutch wastewater treatment plant
provided hypotheses for interactions of anammox bacteria and other nitrogen cycling organisms in these important systems.
Ongoing work in this area includes environmental metagenomics of extreme environments, such as a hydrothermal vent system and an alkaline soda lake.
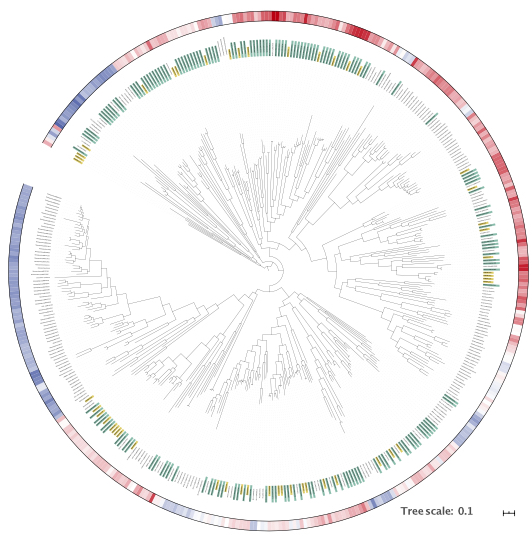
Marker gene mining
I use the phrase “marker gene mining” to mean searching for (metagenome) sequencing reads matching a specific protein family,
for example a marker for a certain metabolic process. I have used this method to study the mcrA gene,
which encodes the large subunit of the methyl-coenzyme M reductase, the last step of methanogenesis
(or the first of methane oxidation using the same pathway). That study was useful to gain insight in global mcrA diversity, but in hindsight I
should have included sequencing data from the IMG repository, because that is where the most interesting novel data was,
as shown later by Borrel and colleagues
and Wang and colleagues.
Marker gene mining can be done using HMMs, if a good representation of the diversity of a gene family is available, or using sequence homology coupled with
false positive filtering using a BLAST score ratio (BSR) approach. The latter is my preferred method,
which I developed for this work, based on the comparative genomics work of
Rasko and colleagues. For a sequencing read, determine the homology score
to the protein family of interest, and to a large set of sequences used as outgroup (such as the NCBI-nr protein database).
If the read has better homology to a sequence in the outgroup set than in the protein family set, its BSR will be (substantially) lower than one.
This allows for easy screening of true positives, regardless of the score of the original hit. The major caveat is that sufficient
homology needs to be present in the sequencing read, so this method might run into trouble with very divergent gene variants.
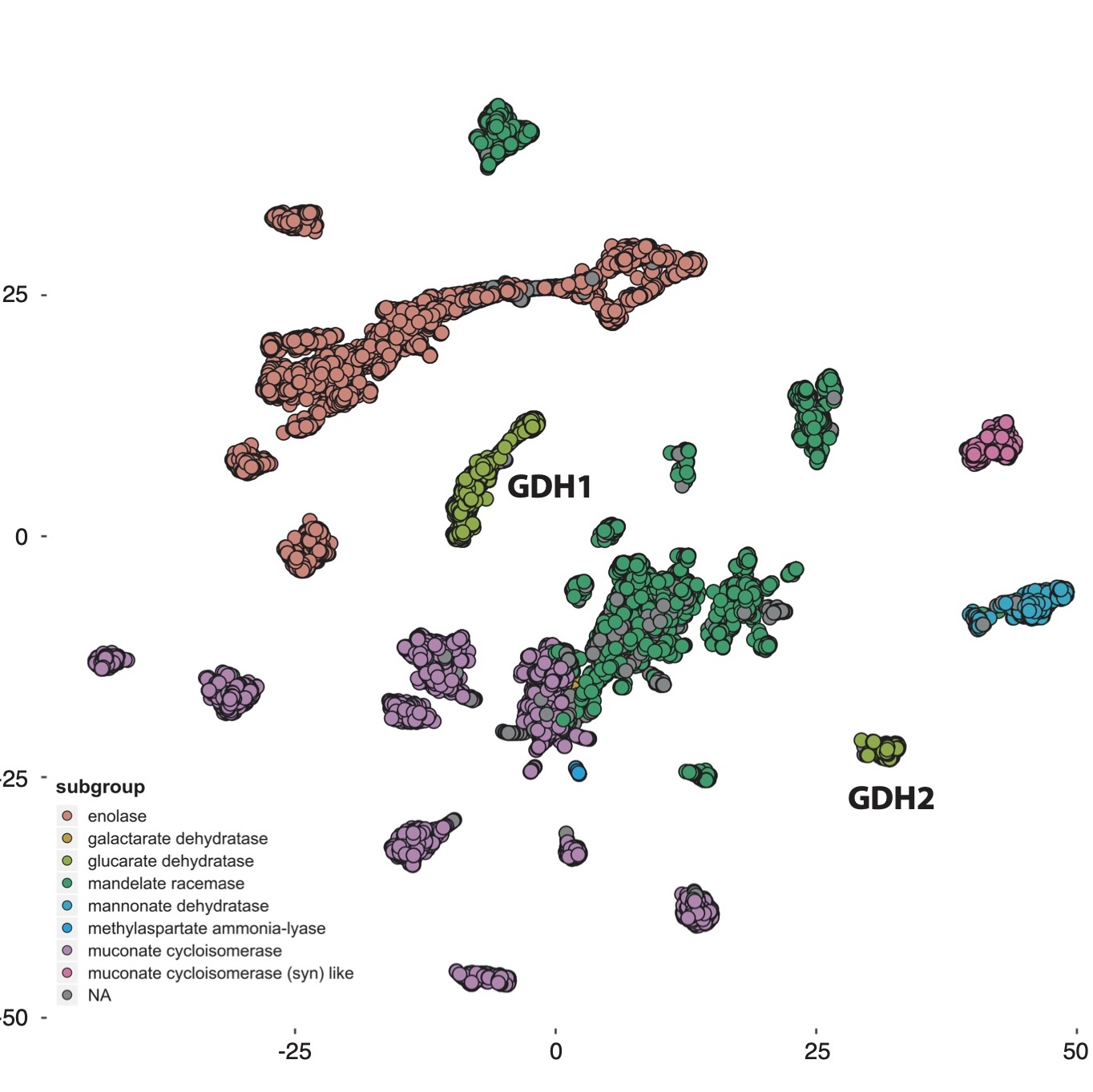
ASM-clust: Protein superfamily analysis
With increased availability of genome information across the tree of life, we get more and more information on the variation within protein families
as well. This allows us to study the evolutionary relationships of complex protein superfamilies that encompass proteins with many different functions.
In most cases, only a few members of complex, functionally diverse, superfamilies have been biochemically characterized. Using a classification
approach on the ever increasing amount of protein sequence informaton, we can identify where the gaps in our knowledge are, and use that information
to maximize impact of more challenging and time-consuming biochemical analyses. To this end, I’ve developed
ASM-clust, an approach to classify protein families based on a matrix of pairwise alignment scores.
The apporach builds on the sequence similarity network (SSN) method
developed in Prof. Patrica Babbitt’s group, but rather than using all possible pairwise alignments, ASM-clust calculates only a subset of alignments and
compares the resulting alignment score profiles (i.e. similarity in multi-dimensional space). I’m excited about this, because the results can be visualized
easily without specific software, i’ve tested it on protein families of 100,000+ sequences, and it handles multi-domain proteins well. I consider
elucidating the biochemistry of proteins of unknown function one of the great challenges of our time, as this functional diversity is a treasure trove
of biotech potential, and I hope ASM-clust will contribute a little to progress in this area!